Project 25:
Bayesian Optimized De Novo Drug Design for Selective Kinase Targeting
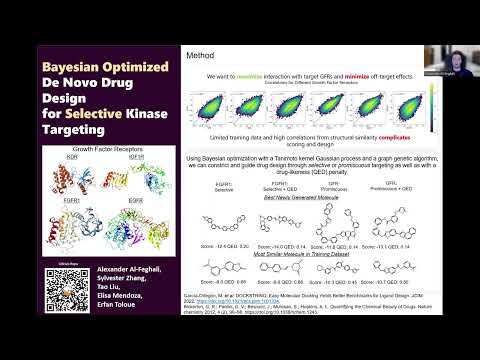
This project employs Bayesian optimization using Gaussian process (GP) models with the Tanimoto kernel and fingerprint features for de novo design of selective growth factor receptor (GFR) inhibitors. GP surrogate models of docking scores will drive optimization of a docking-based objective function balancing potent target binding and minimal off-target interactions, with a drug-likeness penalty. The GP is trained on a subset of data, with new molecules proposed by a graph genetic algorithm, scored, and used to retrain the GP iteratively, aiming to identify promising selective GFR inhibitor candidates.
References:
- Bickerton, G. R.; Paolini, G. V.; Besnard, J.; Muresan, S.; Hopkins, A. L. Quantifying the Chemical Beauty of Drugs. Nature chemistry 2012, 4 (2), 90–98. https://doi.org/10.1038/nchem.1243.
- García-Ortegón, M.; Simm, G. N. C.; Tripp, A. J.; Hernández-Lobato, J. M.; Bender, A.; Bacallado, S. DOCKSTRING: Easy Molecular Docking Yields Better Benchmarks for Ligand Design. Journal of Chemical Information and Modeling 2022, 62 (15), 3486–3502. https://doi.org/10.1021/acs.jcim.1c01334.